Introduction
Robin Buell and collaborators recently published an article in which they used IGB to visualize data from RNA-Seq analysis of various reproductive tissues and stages from corn (Zea mays variety B73) and showed a number of interesting patterns with respect to gene expression. For your convenience, the article is attached to this wiki page. Note the authors used Integrated Genome Browser to create Figure 4.
- Download a copy of the paper: PDF
I wanted to make these new data available to IGB users via the QuickLoad mechanism, but when I started this project, the latest version of the Zea mays B73 genome was not yet available in our main IGB QuickLoad site.
This page describes how I went about obtaining the genome sequence data and foundation gene annotations, reformatted them as needed, deployed them on our main QL site (igbquickload.org) and also how I processed the RNA-Seq data generated in the study and made it available for visualization in IGB.
Methods/Results
Defining the genome
After some Web searches and corresponding with scientists at Phytozome (who directed me to maizesequence.org), I found what appeared to be the authoritative site for maize sequence data at http://www.maizesequence.org. From there, I located the latest maize genome sequence and downloaded it. I also found a single "GFF" file that appeared to contain the latest maize gene structure annotations. I downloaded these, as well.
I downloaded the sequence data from: http://ftp.maizesequence.org/current/assembly/. Within this directory I found files for each chromosome, plus a big file that appeared to contain all of the sequences in a single tarred archive file.
I learned from corresponding with the maize sequence annotators in Doreen Ware's lab (her lab develops Gramene and other key plant bioinformatics resources) that the most up-to-date release of the maize genome is named RefGen_v2 and that it was released in March 2010. So I decided that our QL site would release this genome version as Z_mays_B73_Mar_2010.
The latest gene structure annotations were released more recently and are called the 5b release and are available as the file ZmB73_5b_FGS.gff.gz in GFF format.
After downloading and unpacking the sequence file onto my Mac laptop, I used Unix cat to assemble them into a single file and then used Unix grep to get the fasta headers for all the sequences:
>chromosome:AGPv2:1:1:301354135:1 chromosome 1 >chromosome:AGPv2:2:1:237068873:1 chromosome 2 >chromosome:AGPv2:3:1:232140174:1 chromosome 3 >chromosome:AGPv2:4:1:241473504:1 chromosome 4 >chromosome:AGPv2:5:1:217872852:1 chromosome 5 >chromosome:AGPv2:6:1:169174353:1 chromosome 6 >chromosome:AGPv2:7:1:176764762:1 chromosome 7 >chromosome:AGPv2:8:1:175793759:1 chromosome 8 >chromosome:AGPv2:9:1:156750706:1 chromosome 9 >chromosome:AGPv2:10:1:150189435:1 chromosome 10 >chromosome:AGPv2:mitochondrion:1:569630:1 chromosome mitochondrion >chromosome:AGPv2:chloroplast:1:140384:1 chromosome chloroplast >chromosome:AGPv2:UNKNOWN:1:7140151:1 chromosome UNKNOWN
I also used Unix to parse out the names of sequences represented in the GFF file, like so:
pi$ gunzip -c ZmB73_5b_FGS.gff.gz | cut -f 1 | sort | uniq 1 10 2 3 4 5 6 7 8 9 Mt Pt UNKNOWN
The tools I typically use to convert fasta sequence into IGB-compatible data formats would use the fasta headers to name the sequences. Because I want the sequence names to match chromosome names listed in the GFF annotations file, I decided to make a new fasta file that used the much simpler, more intuitive sequence names from the GFF files instead. So wrote a simple python script to do this (fixMaizeChromNames.py) and checked it into a Loraine lab publicly available subversion repository https://svn.transvar.org/repos/genomes/trunk/pub/src.
Note: The genomes subversion repository is quite large - it contains many data files, including sequence and annotations, for many genomes. Mostly these include plant genomes, in large part because other QuickLoad sites hosted elsewhere are doing a great job thus far of supporting animal and fungal genomes.
This means that if you want to get just the source code, you should check it out like so:
svn co https://svn.transvar.org/repos/genomes/trunk/pub/src genomes_src
This will check out just the source code directory (and Tests) into a new directory called genomes_src. (You can use anything in place of genomes_src if you like – if you don't include anything, then the new directory will be called "src," which is not as useful a name.)
Using this script, I created the new fasta file. Next, I used Jim Kent's faToTwoBit file to create the maize genome sequence file Z_mays_B73_Mar_2010.2bit. I then used his twoBitInfo file to create a genome.txt file that reports the names and sizes of all the sequences represented in the 2bit file:
pi$ faToTwoBit Z_mays_B73_Mar_2010_fixed.fa Z_mays_B73_Mar_2010.2bit pi$ twoBitInfo Z_mays_B73_Mar_2010.2bit genome.txt
Note that the 2bit format (developed by Jim Kent at UCSC) allows IGB to retrieve sections of the data via partial HTTP access. When users request genomic sequence data, IGB will make a request to the server to retrieve just the part of the sequence it needs.
I wrote the UCSC Genome Bioinformatics help list to get the most up-to-date version of these tools. For your convenience, I re-post the reply here:
Hi Ann,
These programs are available for download here:
http://hgdownload.cse.ucsc.edu/admin/exe/. To see the usage statement,
run the program with no arguments.
Please contact us again at genome@soe.ucsc.edu if you have any further
questions.
Regards,
---
Luvina Guruvadoo
UCSC Genome Bioinformatics Group
Next, I wrote a GFF parser to convert the GFF file into BED format. The file is called maizeGffToBed.py and I checked it into subversion under https://svn.transvar.org/repos/genomes/trunk/pub/src.
Using a text editor, I created an annots.xml file for this new genome and its annotations:
<files>
<file name="Z_mays_B73_Mar_2010.bed.gz"
title="Maize Transcripts 5b release"
description="Annotations from MaizeSequence.org, filtered gene set 5b release"
url="http://www.maizegenome.org"
load_hint="Whole Sequence"
label_field="id"
/>
</files>
Note that the load_hint attribute is set to Whole Sequence, which means that when IGB accesses this genome version, it will automatically load the entire set of gene annotations. This is an important feature of IGB; we know from many years of user feedback that the users want to see all the genes as soon as they load a genome.
Finally, I used svn mkdir to create a new subdirectory for this genome in the subversion repository we are using to share and distribute data; its address is https://svn.transvar.org/repos/genomes/trunk/pub/quickload. The new directory is called Z_mays_B73_Mar_2010 and contains the new bed file, the new 2bit sequence file, and the genome.txt file. I then logged onto igbquickload.org (hosted at UNC Charlotte as of October 2011) and ran the command svn up to deploy the new data files. To ensure that IGB would be able to display the new genome version in the IGB Data Access tab, I added the new genome to the contents.txt file in the root QuickLoad directory.
Specifying linkouts
Displaying the maize gene models is great, but this doesn't help users unless they can go from looking at gene structures to finding more information about them. For this, I needed to specify a new "linkout" pattern that would allow IGB to create hyperlinks linking the gene models in the display to external Web pages.
After looking around the maizegenome.org Web site, I realized that all gene models have their own report page at the site and that IGB can link to those pages by constructing links that look like this:
http://www.maizesequence.org/Zea_mays/Transcript/Transcript?t=NAME
where NAME is the name of the transcript, which comes from the name field in the bed file I created from the original GFF file I downloaded from the maizegenome.org site.
Also, I realized that Phytozome has pages for some if not all maize transcripts and that I could "link out" to these pages using this URL pattern recommended by one of the Phytozome developers:
So I decided to implement these link out patterns in IGB. For this, I added the following lines to the igb_default_prefs.xml file (I edited my checked-out copy of the IGB 6.6 branch to start with):
<annotation_url annot_type_regex=".Maize Transcripts." name="MaizeSequence.org" url="http://www.maizesequence.org/Zea_mays/Transcript/Transcript?t=$$" /> <annotation_url annot_type_regex=".Maize Transcripts." name="Phytozome.org" url="http://www.phytozome.net/genePage.php?crown&method=0&search=1&searchText=$$&detail=1" />
To test that the links were working, I next had to delete the file weblinks.xml in my "dot-igb" directory (~/.igb) in order to remove any old patterns saved from previous versions of the preferences file:
pi$ rm ~/.igb/weblinks.xml
Note: Starting with IGB release 6.7, scheduled for January 2012, users won't need to remove weblinks.xml when developing a new linkout pattern. Also, IGB 6.7 will make it possible for individual QuickLoad sites to distribute their own linkout patterns, which is useful for data providers who have developed companion Web sites describing their gene annotations.
I then built the IGB 6.6 branch code, launched the IGB, and then visited the Z_mays_B73_Mar_2010 genome. After the transcripts loaded, I right-clicked one of the newly loaded transcripts to check that the links I specified were there and that they led to the expected site.
Of course, one complication with this is that in order for all users to pick up a new linkouts pattern, the IGB developers need to release a new version of the igb_default_prefs.xml file hosted on the main BioViz IGB deployment site. We don't have to release an all-new build of IGB, but adding new linkout patterns does require updating the file igb_default_prefs.xml that all running IGB instances will access upon startup.
Adding NGS data
The paper I mentioned in the Introduction section reported a Short Read Archive study accession of SRP006463 for all the data sets described in the paper.
Retrieving the data from the Short Read Archive using wget
I looked at the SRA Web site and learned that the SRA organizes sample files under directories named for the study. Each NGS data file is distributed as an ".sra" format file named for the sample type. Furthermore, one can find out information for each file by visiting links that follow this pattern:
http://www.ncbi.nlm.nih.gov/sra?term=NAME
where NAME is an SRA accession, such as SRR189760. Note that in this case, the first three letters "SRR" indicate that this is a "reads" file and contains sequence and sequence quality information.
I then used wget to obtain all the sample "SRR" files for this data set, running a shell script I wrote called getSRP006434.sh:
$ more getSRP006463.sh #!/bin/sh # how I got the maize RNA-Seq data # see: # http://www.ncbi.nlm.nih.gov/sra/SRX058606?&report=full # http://en.wikipedia.org/wiki/FASTQ_format wget -nd -nH -r -P SRP006463 ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP006/SRP006463
Note that this command will put all files it finds under this study directory into an output directory named SRP006463 and all the files will be at the same level.
Converting SRA files to fastq using fastq-dump
Once I had downloaded all the files, I then used the program fastq-dump (from the SRA toolkit - I used version named sratoolkit.2.1.6) to convert each '.sra' file to a .fastq file, like so:
$ fastq-dump *.sra
Of course, many of these commands take relatively long periods of time to complete. Since I was doing most of this on a remote server, I needed to ensure that if my connection to the server was lost, the command would continue and not be killed. To ensure this would not happen, I used a useful program called screen to create a new shell and then used that shell to run longer-running commands.
Running tophat to align the reads on the genome
Once the files had been converted, I then used a simple shell script to run TopHat, an alignment program for short reads data, like so:
#!/bin/sh
O=tophat_5
FILES=/afs/transvar.org/projects/illumina/SRA/SRP006463/*.fastq
B=$HOME/data/bowtieindex/Z_mays_B73_Mar_2010
for F in $FILES
do
T=${F##*/}
S=${T%.fastq}
C="tophat --segment-length=17 -p 30 -I 8000 -o $O/$S $B $F"
echo "Starting $C"
$C
done
Before I wrote the script, I did a quick analysis of the intron sizes in the maize genome and found that the vast majority (more than 99%) are smaller than 8000 bases. Thus, I choose a value of 8000 for the -I (maximum intron size) parameter. Also, before I ran TopHat, I first had to create a bowtie index for it. For this, I used the modified genome fasta file I had used previously to create the twoBit file Z_mays_B73_Mar_2000.2bit.
I ran this script on a Dell 910 processing server equipped with 512 Gig of memory and 32 processors; Each data file required about 1 hour to process, which is more or less typical for these kinds of data sets. (Many thanks to Dr. Xiuxia Du's lab here at the North Carolina Research Campus for allowing me to use their server.)
Next, I processed the data using a custom program that uses samtools and a modified version of the wiggles program (from a TopHat distribution) to generate data files for visualization in IGB. These included sorted, indexed BAM files contain reads that map exactly once to the reference (extension sm.bam) and BAM files that contain reads that map multiple times to the reference (extension mm.bam) along with bedgraph (wiggle) format files (from the wiggles program) that report coverage across the reference. It also performs some re-formatting and renaming of the junction file TopHat produced.
A compiled version of the wiggles program is available for Mac and Linux as an attachment.
Because the wiggles program is not widely known, I'll describe how to use it here.
Using wiggles to make a bedgraph coverage graph files from a BAM alignments file
The modified wiggles file accepts SAM format on stdin and emits bedgraph format data on stdout. For example, to create a bedgraph file called test.bedgraph, you would do this:
$ samtools view test.bam | wiggles.linux > test.bedgraph
Compressing and deploying the files
Finally, I compressed the "bed" and "wig" files in the output directory and then logged into the machine that hosts our public QuickLoad site http://www.igbquickload.org. From there, I used rsync to copy the files from the processing server onto the quickload host.
To deploy the files, I wrote a script in python annots_xml_for_SRA.py that generates annots.xml formatted text describing the different files. I then copied and pasted (by hand) the XML into the annots.xml file for the Z_mays_B73_Mar_2010 genome data directory.
Using tabix to create a sorted, random-access wig file
Upon testing the files in IGB, I realized that the "bedgraph" files are very slow to load. At our weekly group meeting, the IGB developers suggested I use the tabix utility to convert the wig files to random access "tabix" format which IGB now supports starting with the 6.6 release.
Note however that IGB 6.6 supports tabix for files with extension bedgraph. The wig format, as I have been using it, is also bedgraph format, consisting of a sequence name (column 1), a start position (column 2), an end position (column 3), and a numeric value (column 4). However, for IGB to recognize a "tabixed" wig file, I needed to give it the extension "bedgraph."
For more information about tabix and how to use it, see this paper by Heng Li.
To try this out, I first created a simple test file like so:
$ gunzip -c SRR189770.sm.wig.gz | grep -v track > test.bedgraph $ grep ^UNKNOWN test.bedgraph | head -n 1000 > littlewig.bedgraph $ grep ^1 test.bedgraph | head -n 1000 >> littlewig.bedgraph $ grep ^10 test.bedgraph | head -n 1000 >> littlewig.bedgraph $ grep ^5 test.bedgraph | head -n 1000 >> littlewig.wig
Next, I sorted it like so, following documentation I found here.
sort littlewig.bedgraph -k1,1 -k2,2n > littlewig.sorted.bedgraph
Note that the "n" character forces a numeric sort on the start column of the bedgraph file. The primary sort (-k1,1) is not a numeric sort, however. Translating into English, I first wanted to sort on field one through field one (-k1,1) and then wanted to sort numerically on the start position, field two through field two (-k2,2n).
Following this, I then compressed and indexed the small file, like so:
$ bgzip littlewig.sorted.bedgraph $ tabix -s 1 -b 2 -e 3 littlewig.sorted.bedgraph.gz
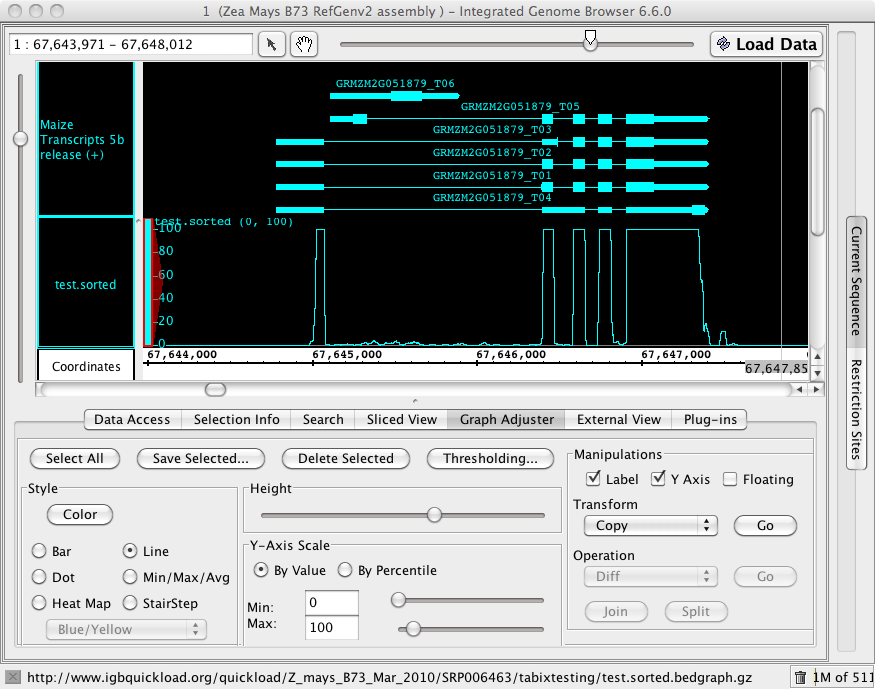
To test whether this new file could be read into IGB, I made a new directory under Z_mays_Mar_2010_B73/SRP006463 called tabixtesting. I then launched IGB 6.6 (the released version) and click-dragged the file http://www.igbquickload.org/quickload/Z_mays_B73_Mar_2010/SRP006463/tabixtesting/littlewig.sorted.bedgraph.gz directly into the IGB interface. And it worked! I was able to view data.
Next, I decided to test a much larger file - test.bedgraph which contains a lot more data: 328 Mb.
I launched a screen session and from this new shell, ran these commands:
$ sort test.bedgraph -k1,1 -k2,2n > test.sorted.bedgraph & $ bgzip test.sorted.bedgraph & $ tabix -s 1 -b 2 -e 3 test.sorted.bedgraph.gz &
To test, I opened the Web directory http://www.igbquickload.org/quickload/Z_mays_B73_Mar_2010/SRP006463/tabixtesting and click-dragged the new file test.sorted.bedgraph.gz into IGB.
I then tested loaded some regions into the viewer. The speed was much better than the "wig" files I created originally.
Conclusion
Using tabix-processed bedgraph files to distribute wiggle files (coverage graphs) is a viable solution for sharing data from maize RNA-Seq experiments. However, I will need to modify my procTopHat.py script to include bgzip compression and tabix indexing.
Figure 1. An image from IGB showing a genome bedgraph data file partially loaded onto chromosome 1 of the maize genome.